| QMS那回事 | 您所在的位置:网站首页 › 制定制订的区别 标准 › QMS那回事 |
QMS那回事
|
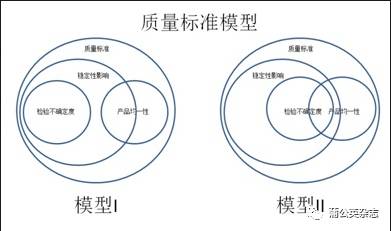
1.4 OOS和OOT OOS是指检验结果超出质量标准的规定。是一种发生在检验中的偏差,有严谨程序性的调查过程。 并不是所有超出标准都按照OOS的程序进行调查,典型的例如中间控制。 OOT是指检验结果超出正常的趋势。这种趋势可能通过统计分析得出,可以与警戒限/行动限关联,但并不一定关联。 OOT与警戒限/行动限的区别: OOT的目的是为了确认检验结果的可靠性和调查发生趋势偏离的原因; 警戒限/行动限是为了避免系统发生偏离和不符合。 有时宁可错杀不能放过! 2 检验的目的 为什么说检验目的?因为检验目的直接决定了是否制定内控标准。检验目的主要有两种:确认符合性和指导生产、放行,前者我们没有必要制定内控标准,后者我们就需要考虑制定内控标准了。 2.1 确认符合性 确认产品/物料/中间产品/公用介质等符合要求,重要的是结论,而不是结果。包括: Ø 鉴别 Ø 限量法的杂质检查,例如入厂API的氯化物、重金属等 Ø 一些非关键的质量属性 ,例如入厂API的残留溶剂(对于生产就不同了)、API有关物质中的工艺杂质 Ø 不影响制剂产品CQA的物料的质量属性,例如用于注射剂的API的粒度分布 2.2 指导生产、放行 确保结果对产品和生产的影响符合预期,或在控制的范围之内。包括: Ø CQA检验项目,例如降解物 Ø 影响产品CQA的物料/中间产品/公用介质的检验项目,例如用于固体制剂的API的粒度分布 3 制剂产品内控标准 3.1 目的 确保产品在生命周期里质量持续符合预期和质量标准的规定,以确保患者用药的安全性和有效性。 3.2 影响产品生命周期内符合性的风险源有哪些?这里面就包括了: Ø 内控标准 Ø 产品稳定性 Ø 产品均一性(批内产品个体差异,即生产能力) Ø 检验的不确定度(单次检验差异,即检验能力) 下面是两个质量标准的模型:
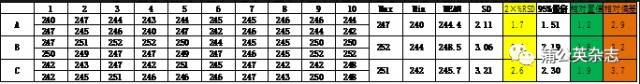
3.3 解释一下: Ø 质量标准(产品符合性标准)应考虑稳定性、产品均一性和检验不确定度的影响。 Ø 稳定性检测数据中已经蕴含了检验不确定度;由于取样数量少代表性差,产品均一性可能会、但并不一定会影响稳定性数据。 Ø 根据检验方法不同,产品均一性可能会影响(例如投片法制备样品)单次检验的不确定度,也可能不影响(例如研磨法制备样品)。 Ø 当稳定性没有发生变化或变化较小时,影响质量标准的主要因素只有检验不确定度和产品均一性。 3.4 制订内控标准的原则 Ø 非CQA项可以不需要制定,使用国家标准即可。 Ø 对于稳定性较差的项目,应综合考虑稳定性(其中已经包括了检验不确定度)和产品均一性的影响。 Ø 对于稳定性较好的项目,可以综合考虑检验不确定度和产品均一性的影响。 3.5 含量内控标准的建立 3.5.1 三种计算方法 Ø 标准偏差法:2RSD(目的是为了折算到100%) Ø 相对偏差法:(最大值-最小值)/平均值 Ø 95%置信限法: 3.5.2 检验不确定度 同一批产品不同检验员测定的结果 3.5.3 产品均一性 不同批次产品间片重的差别[20片平均片重(mg)]
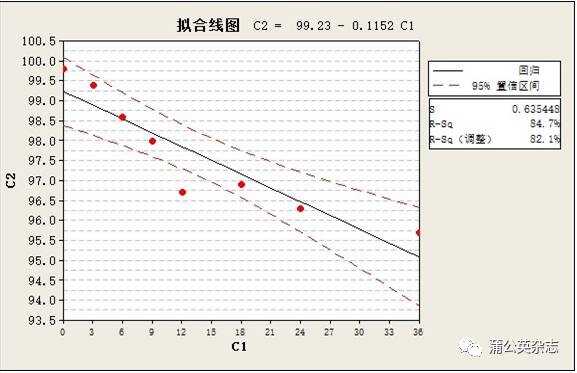
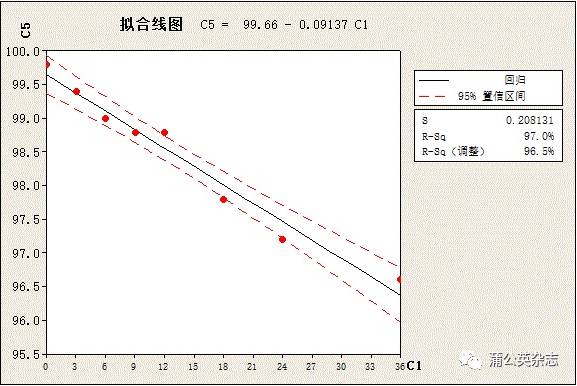
3.5.4 稳定性 不同批次产品稳定性测定的结果
不同批次产品稳定性测定的结果(修正片重)
3.5.5 含量内控标准下限 Ø 存在稳定性下降趋势的产品 内控下限=标准下限+折片重稳定性下降值+均一性 例:以95%置信限计算上述最差结果 内控下限=90.0%+3.8%+1.9%=95.7% Ø 不存在稳定性下降趋势的产品 内控下限=标准下限+不确定度+均一性 例:以95%置信限计算上述最差结果 内控下限=90.0%+0.8%+1.9%=92.7% 3.5.6 含量内控标准上限 内控标准上限=2T-LRT 式中LRT为内控标准下限 T为目标含量,按照药典要求理论为100% 中国的要求:含量在贮存期有下降趋势的,可以适当增加投料量,即T>100%。 3.5.7 含量内控标准的注意事项 Ø 内控标准与趋势限度:趋势限度体现的是生产控制能力,应在内控标准范围内。 Ø 内控标准与OOS:超标时放行、拒绝? Ø 内控标准与警戒限/行动限:IPC片重检查可以设定行动限,超限重新调片重。 Ø 不折算片重/装量的检验方法,检验不确定度中已经包含了产品均一性。 3.6 有关物质内控标准的注意事项 Ø 自身对照法时,产品均一性影响几乎可以忽略不计;而外标法则必须要考虑。 Ø 检验不确定度重点考虑仪器间的差异,而不是进样差异和人员差异,尤其是对于低限度标准的杂质。 Ø 关注降解物,对于API工艺杂质不需要考虑。 Ø 只有标准上限,杂质95%置信限曲线应采用上侧线。 4 制剂原辅料内控标准 4.1 目的 确保使用受到控制的物料能够持续生产出符合预期质量的制剂产品。 所需控制的是,那些可能直接影响制剂CQA的CMA。 影响制剂CQA的因素:CMA、CPP、环境、包装、贮存。 4.2 物料API的内控 4.2.1 有关物质: 降解物:应确保生产出的制剂放行标准,需要考虑API的贮存,有一个客户标准,必要时再有一个厂内的复验标准。 工艺杂质:除非制剂有相应要求,已知的、未知的或总杂质,没有必要设置内控标准。 未知杂质:应不是降解物,确保制剂的未知杂质和总杂质。 4.2.2 外观 应考虑API本身的颜色对制剂的影响,尤其是液体制剂。必要时可以用UV-Vis控制。 4.2.3 晶型 一般药典/国家标准不控制,固体制剂可能影响BE和体外溶出度。控制方法:粉末X射线衍射、熔点、红外等。必要时应使用专属性强的方法。 4.2.4 粒度分布 一般药典/国家标准不控制,固体制剂可能影响BE和体外溶出度。控制方法:激光粒度、筛分法。 4.2.5 含量测定 较窄的范围没有必要内控,例如98.0%~102.0%。 4.3 辅料的内控 重点在于功能性控制,例如填充剂的粒度分布、粘合剂的黏度、缓冲剂pH值等。 特殊——十二烷基硫酸钠: 十二烷基比例差别大(含辛烷基、癸烷基、十二烷基、十四烷基等),可能影响溶出曲线。 5 制剂/API中间产品质量标准 5.1 目的 1)确保使用受到控制的中间产品能够持续生产出符合预期质量的API/制剂产品。 2)能够反映工序的重现性。 所需控制的是,那些可能直接影响API/制剂CQA的质量属性。 5.2 API中间产品的控制 5.2.1 有关物质 关键是特定杂质的控制 Ø 本工序和后续工序中难以去除的杂质; Ø 在传递后生成的杂质不易除去的杂质; Ø 影响后续工序收益的杂质; 5.2.2 含量 外标法含量测定与否取决于工艺和成本的需求,并不是必须的。 5.2.3 残留溶剂 确保所关注的残留溶剂是否除去,并不需要批批测,可以采用验证加抽检的方式。 5.3 制剂中间产品的控制 5.3.1 含量 应不超过制剂成品的标准范围,含量测定的目的是哪种? Ø 指导生产? Ø 工序确认? 用于指导生产往往结果可能是灾难性的,尤其是成品标准范围较窄的产品。这是由于取样的代表性和检验的不确定度造成的。 5.3.2 有关物质 只有那些在生产过程中CPP可能影响到有关物质的中间产品需要关注,例如工艺中有局浓或者高温降解的情况发生。 5.3.3 其他 Ø 粒度分布 Ø 外观(颜色) Ø 水分 Ø pH …… 6 包装材料内控标准 6.1 与产品稳定性相关 Ø 粘合性能 Ø 密封性能 Ø 厚度/厚度均匀性 Ø 透光性能 Ø 注射剂相容性相关的溶出、脱落物 6.2 与包装工序相关 Ø 规格 Ø 上机性能 6.3 与销售相关 6.3.1 印刷性包材 Ø 色差 Ø 印刷质量 Ø 规格、样式和内容 ——建议加编号和版本号控制 6.3.2 药品电子监管码 Ø 规格(尺寸) Ø 可读性(使用抄码枪可以读出) Ø 读数准确性(读出的数据是正确的) Ø 产品一致性(前七位与申请药监码的一致) 6.3.3 运输牢固性能——产品与包材结合的牢固性 7 制药用水限度 7.1 目的 1)确保持续生产出符合生产能力的制药用水。 2)及时发现不良趋势。 对注射剂来说,那些可能影响制剂的CQA。 7.2 微生物限度 7.2.1 培养基筛选 需要根据水质情况确定选择使用哪类培养基:富营养培养基,还是低营养培养基。 7.2.2 样品量筛选 使用多少量用于检验合适?典型的细菌检测要求是30~300CFU/皿,我们应选择合适的水的数量,使检测结果有统计学意义。 7.2.3 计数 各国药典限度标准的单位都是CFU/ml,如果我们要按照这个标准报告,有很多结果将是“ |
【本文地址】